Interactions médicamenteuses : mécanismes et risques à connaître
Les interactions médicamenteuses représentent l’une des premières causes d’hospitalisations évitables en France : selon les données de pharmacovigilance compilées par l’ANSM (Thésaurus, mis à jour juin 2024), elles sont impliquées dans près de 10 % des effets indésirables graves notifiés chaque année. Pourtant, la grande majorité de ces accidents est prévisible — et donc évitable — à condition de comprendre les mécanismes en jeu. Ce guide vous explique comment deux médicaments pris en même temps peuvent, silencieusement, se neutraliser ou au contraire s’amplifier mutuellement, et quelles combinaisons surveiller en priorité.
Que vous preniez un traitement au long cours ou que vous ajoutiez un médicament en automédication, les informations qui suivent vous donneront les clés pour dialoguer efficacement avec votre pharmacien.
📑 Sommaire de l’article
- 1. Qu’est-ce qu’une interaction médicamenteuse ?
- 2. Interactions médicamenteuses : quatre niveaux de risque selon l’ANSM
- 3. Interactions pharmacodynamiques : quand les effets s’additionnent ou s’annulent
- 4. Interactions pharmacocinétiques : le rôle central des cytochromes P450
- 5. Interactions médicamenteuses majeures à connaître au comptoir
- 6. Anticoagulants oraux directs (AOD) : un profil d’interactions médicamenteuses à part
- 7. Millepertuis : la plante aux cent interactions médicamenteuses
- 8. Prévenir les interactions médicamenteuses : les réflexes qui sauvent
1. Qu’est-ce qu’une interaction médicamenteuse ?
On parle d’interaction médicamenteuse lorsque la présence simultanée de deux substances dans l’organisme modifie l’effet de l’une ou de l’autre — voire des deux. La modification peut aller dans deux sens opposés :
- Insuffisance d’effet : l’un des médicaments est métabolisé trop vite, absorbé en quantité insuffisante, ou bloqué dans son action — il ne remplit plus son rôle thérapeutique. C’est ce qui arrive, par exemple, lorsqu’une patiente sous contraceptif oral commence un traitement par millepertuis.
- Excès d’effet ou toxicité : l’un des médicaments s’accumule anormalement dans l’organisme, ou ses effets sont amplifiés par l’autre — avec un risque d’effets indésirables graves, parfois engageant le pronostic vital. C’est le cas classique de l’association AVK + AINS (anti-inflammatoires non stéroïdiens), qui peut provoquer une hémorragie sévère.
ℹ️ Un chiffre à retenir
Le rapport d’activité 2024 de l’ANSM recense 949 cas marquants de pharmacovigilance ayant conduit à 1 062 mesures de réduction du risque sur la seule année 2024. Les interactions médicamenteuses figurent parmi les causes les plus fréquentes de ces signalements. En pratique officinale, c’est au minimum une dispensation sur deux en polypharmacie qui mérite une vérification active des interactions.
👨⚕️ Conseil au comptoir
Deux médicaments pris à plusieurs heures d’intervalle peuvent tout de même interagir, car leurs concentrations plasmatiques (la quantité de principe actif circulant dans le sang) se chevauchent dans le temps. L’espacement des prises réduit parfois le risque, mais ne le supprime jamais complètement pour les substances à demi-vie longue.
2. Interactions médicamenteuses : quatre niveaux de risque selon l’ANSM
Le Thésaurus des interactions médicamenteuses de l’ANSM — dont la dernière mise à jour majeure date de septembre 2023, avec un différentiel publié en janvier 2024 — classe chaque interaction en quatre niveaux de contrainte croissante, du plus souple au plus strict.
| Niveau | Signification clinique | Conduite à tenir | Niveau de preuve ℹ️ |
|---|---|---|---|
| Contre-indication (CI) | Risque grave documenté, bénéfice-risque défavorable dans tous les cas | Association absolument à ne pas réaliser | ⭐⭐⭐⭐⭐ |
| Association déconseillée (AD) | Risque réel, l’association doit être évitée sauf nécessité absolue | Évaluation bénéfice-risque individuelle, surveillance rapprochée si maintenu | ⭐⭐⭐⭐ |
| Précaution d’emploi (PE) | Association possible à condition de respecter les recommandations posologiques et de surveillance | Adaptation de dose, surveillance clinique et/ou biologique | ⭐⭐⭐ |
| À prendre en compte (APEC) | Risque faible ou potentiel, difficilement quantifiable | Information du prescripteur, vigilance clinique | ⭐⭐ |
👨⚕️ Conseil au comptoir
La distinction entre « contre-indication » et « association déconseillée » est fondamentale : la première est absolue (elle ne se discute pas), la seconde peut être maintenue sous surveillance étroite si le bénéfice clinique l’emporte. C’est tout le travail du pharmacien clinicien que d’évaluer ce curseur pour chaque patient.
3. Interactions pharmacodynamiques : quand les effets s’additionnent ou s’annulent
Les interactions pharmacodynamiques (du grec dunamis, « force ») surviennent quand deux médicaments agissent sur le même système physiologique — sans nécessairement se gêner dans leur absorption ou leur métabolisme. Imaginez deux personnes qui poussent la même porte en même temps : l’effet est multiplié, parfois dangereusement.
Synergie et addition d’effets indésirables
Les exemples les plus fréquents en pratique officinale :
- Addition d’effets bradycardisants : bêtabloquants + inhibiteurs calciques bradycardisants (vérapamil, diltiazem) + digitaliques + inhibiteurs de la cholinestérase (utilisés dans la maladie d’Alzheimer). Cette quadruple menace sur le nœud sinusal (le « chef d’orchestre » du rythme cardiaque) peut conduire à un bloc auriculo-ventriculaire grave.
- Addition d’effets atropiniques : antihistaminiques H1 de première génération + antidépresseurs tricycliques + neuroleptiques phénothiaziniques. Ces trois classes bloquent toutes le récepteur muscarinique M1, entraînant sécheresse buccale, rétention urinaire, confusion et constipation — surtout sévères chez le sujet âgé.
- Majoration du risque hémorragique : AVK ou AOD + AINS + antiagrégants plaquettaires (aspirine, clopidogrel). Ces molécules perturbent la coagulation par trois mécanismes différents mais additifs, multiplient le risque de saignement digestif, parfois fulminant.
- Toxicité musculaire (rhabdomyolyse) : statines + fibrates. Les statines (inhibiteurs de la HMG-CoA réductase, l’enzyme fabriquant le cholestérol) et les fibrates peuvent tous deux léser les cellules musculaires ; combinés, ce risque est multiplié. La rhabdomyolyse (destruction musculaire massive avec libération de myoglobine dans le sang) peut conduire à une insuffisance rénale aiguë.
Antagonisme : quand un médicament en contre un autre
L’exemple le plus parlant au comptoir est l’antagonisme entre antiparkinsoniens dopaminergiques (lévodopa, agonistes dopaminergiques) et neuroleptiques antiémétiques — le métoclopramide (Primpéran®) en tête. Le premier relance la dopamine dans les circuits nigro-striés (le système moteur déficient dans la maladie de Parkinson) ; le second bloque précisément les récepteurs dopaminergiques D2. C’est une guerre chimique dans les ganglions de la base, que le patient paie en aggravation de ses symptômes moteurs.
⚠️ Antiémétiques et Parkinson : une association contre-indiquée
Métoclopramide (Primpéran®), métopimazine (Vogalène®) et alizapride (Plitican®) sont contre-indiqués chez tout patient parkinsonien traité. En cas de nausées, orienter vers la dompéridone (Motilium®), qui bloque les récepteurs D2 périphériques sans franchir la barrière hémato-encéphalique, ou vers des antiémétiques à mécanisme différent après avis médical.
👨⚕️ Conseil au comptoir
Avant de dispenser un antiémétique en automédication, la question « Avez-vous un traitement pour la maladie de Parkinson ? » est non-négociable. Un simple Primpéran® acheté sans ordonnance peut précipiter une crise akinétique chez un patient fragile.
4. Interactions pharmacocinétiques : le rôle central des cytochromes P450
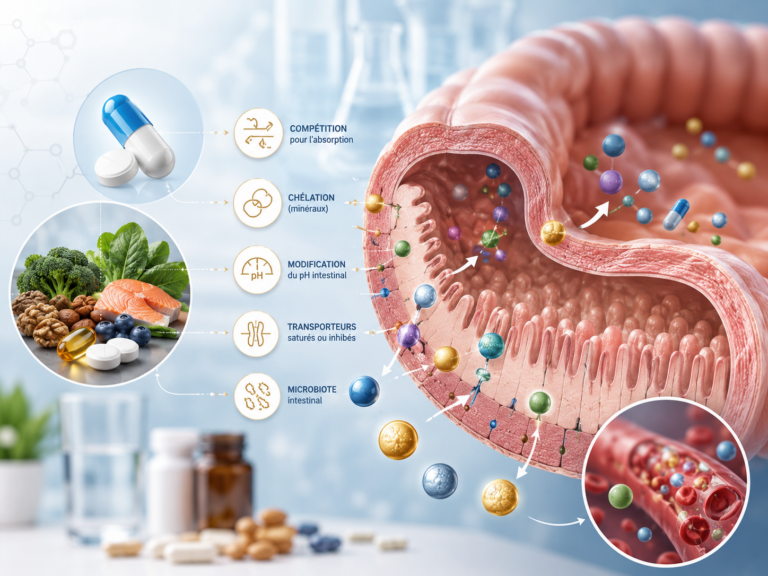
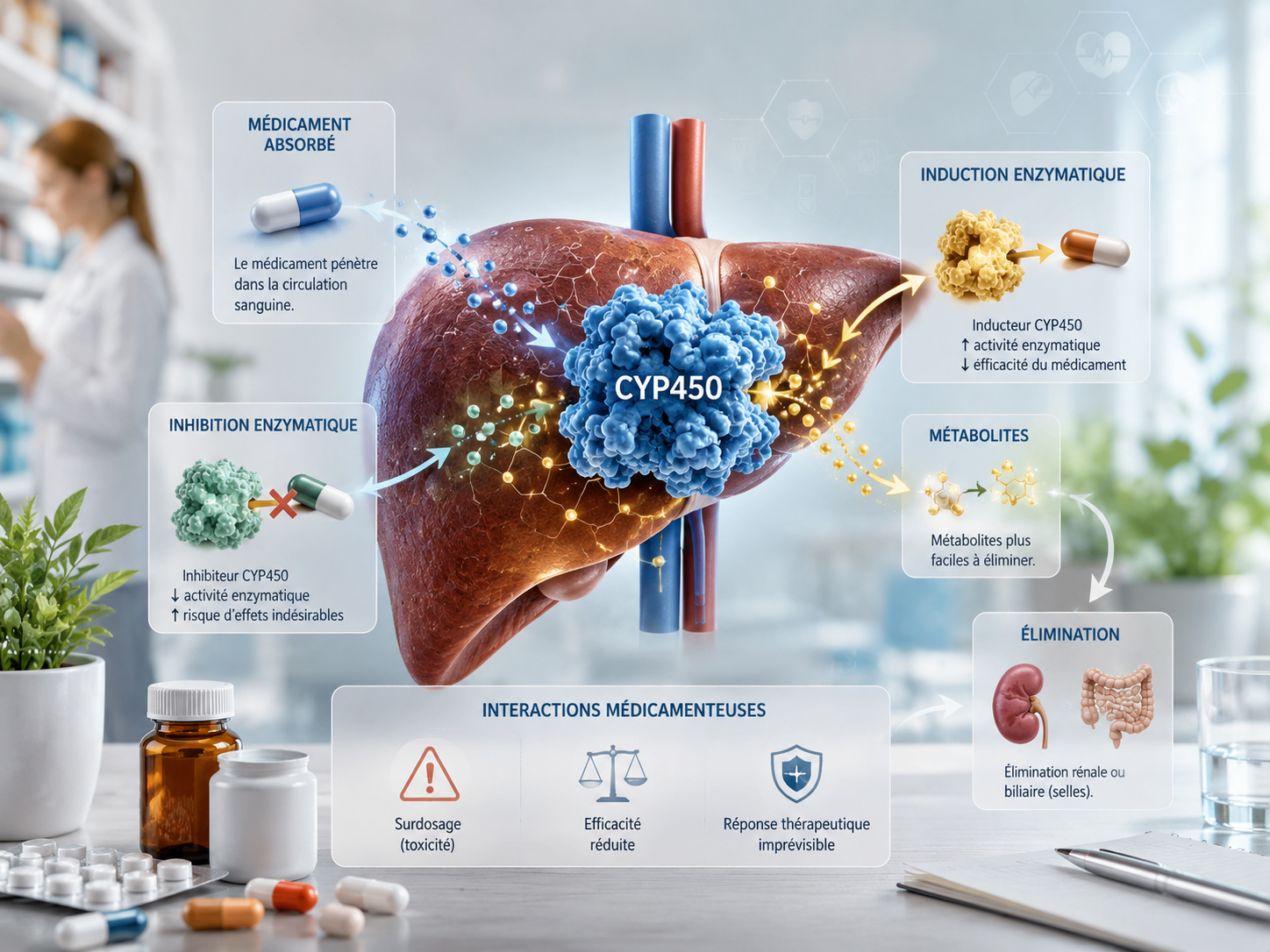
Les interactions pharmacocinétiques — du grec kinêtikos, « qui met en mouvement » — touchent le devenir du médicament dans l’organisme : son absorption dans l’intestin, sa distribution dans les tissus, sa transformation dans le foie, puis son élimination. Chacune de ces étapes est une porte d’entrée possible pour une interaction.
Schéma des interactions médicamenteuses pharmacocinétiques : chaque étape du parcours du médicament dans l’organisme peut être perturbée. Le métabolisme hépatique par les cytochromes P450 (CYP3A4, CYP2D6, CYP2C9) est le site le plus fréquemment impliqué dans les interactions médicamenteuses cliniquement significatives. (Sources : ANSM Thésaurus 2024 ; Deodhar M. et al., Pharmaceutics, 2020)
Le cytochrome P450 : l’enzyme qui change tout
Chez l’homme, le CYP3A4 est quantitativement le plus important des cytochromes P450 : il représente 30 à 50 % du contenu hépatique en enzymes CYP450, avec une présence également au niveau intestinal dans les entérocytes. Environ la moitié de tous les médicaments métabolisés dans l’organisme le sont via le CYP3A4. Un médicament qui « occupe » ou « accélère » cette enzyme peut donc modifier la concentration d’une quantité extraordinairement large d’autres substances.
Les principales substances inhibitrices des cytochromes sont, pour le CYP3A4 : les macrolides (sauf la spiramycine), les antifongiques azolés (fluconazole, itraconazole, kétoconazole), les inhibiteurs de protéase, le cobicistat, l’amiodarone, le vérapamil, le diltiazem, et le jus de pamplemousse. Pour le CYP2D6 : l’escitalopram, la fluoxétine, la paroxétine. Pour le CYP2C9 : les antifongiques azolés et la fluvoxamine. Pour le CYP1A2 : la ciprofloxacine, la norfloxacine, la fluvoxamine.
Les principales substances inductrices (qui accélèrent le métabolisme) sont le millepertuis, la rifampicine, les antiépileptiques (phénobarbital, phénytoïne, carbamazépine), la griséofulvine, certains antirétroviraux (éfavirenz, névirapine), le tabac et l’alcool en consommation chronique.
🔑 Inhibition vs Induction : les deux temps différents
L’inhibition est rapide — elle s’installe en quelques heures dès la première prise, et disparaît à l’arrêt de l’inhibiteur. L’induction, elle, est progressive : l’effet inducteur atteint son maximum en 10 à 15 jours, et disparaît progressivement à l’arrêt de l’inducteur. Conséquence pratique : arrêter brutalement un inducteur peut exposer le patient à un surdosage soudain du médicament substrat dont l’élimination était jusqu’alors accélérée.
La P-glycoprotéine (P-gp) : la deuxième « douane » méconnue
La P-glycoprotéine (P-gp) est une protéine de transport membranaire — imaginez-la comme un portier qui expulse certains médicaments hors des cellules intestinales ou hépatiques avant qu’ils ne passent dans la circulation. La ciclosporine, par exemple, inhibe fortement la P-gp : associée au rivaroxaban, substrat de la P-gp, elle entraîne une augmentation de sa biodisponibilité, donc de son effet anticoagulant. La P-gp est particulièrement importante pour comprendre les interactions des anticoagulants oraux directs (AOD).
👨⚕️ Conseil au comptoir
Quand un patient vous demande si son nouveau traitement peut interagir avec son traitement habituel, deux questions-clés orientent d’emblée : « Est-ce que l’un des deux médicaments est un inhibiteur ou un inducteur du CYP3A4 ? » et « L’un des deux est-il un substrat à marge thérapeutique étroite (anticoagulant, digoxine, ciclosporine, antiépileptique) ? » Si la réponse aux deux est oui — c’est là que se concentrent les vrais risques.
5. Interactions médicamenteuses majeures à connaître au comptoir
Alcaloïdes de l’ergot de seigle : deux contre-indications absolues
Les dérivés ergotés (dihydroergotamine Ikaran®, Seglor® ; ergotamine Gynergène® caféine) sont des vasoconstricteurs puissants utilisés dans la migraine. Leur marge thérapeutique est étroite et leurs interactions, souvent dramatiques :
- + Triptans (almotriptan, élétriptan, sumatriptan, zolmitriptan…) : contre-indication absolue. L’association de deux vasoconstricteurs peut provoquer une crise hypertensive ou une ischémie myocardique ou cérébrale aiguë. Délai à respecter : attendre 24 h après le dernier dérivé ergoté avant un triptan, et 6 h après le dernier triptan avant un dérivé ergoté.
- + Macrolides (érythromycine, clarithromycine, roxithromycine, télithromycine) : contre-indication absolue. Ces antibiotiques sont de puissants inhibiteurs du CYP3A4 — ils bloquent la dégradation des alcaloïdes, dont la concentration plasmatique monte brutalement. Conséquence : ergotisme (vasospasme périphérique, coronaire ou cérébral), parfois avec nécrose des extrémités.
🚫 Ergotés + macrolides : l’interaction qui défigure
Un patient migraineux sous Ikaran® qui débute une angine et reçoit de la clarithromycine (Naxy®, Zeclar®) présente un risque réel d’ergotisme aigu. Toujours alerter le prescripteur et orienter vers un antibiotique non inhibiteur du CYP3A4 (amoxicilline en première intention si le profil bactériologique le permet). La spiramycine (Rovamycine®) est le seul macrolide épargné par cette interaction.
Antiarythmiques et risque de torsades de pointes
L’allongement de l’intervalle QT — la durée de repolarisation ventriculaire mesurable sur l’électrocardiogramme — est le principal mécanisme des arythmies induites par les médicaments. Certaines combinaisons le majorent à un point critique, déclenchant des torsades de pointes (arythmies potentiellement fatales) :
- Antiarythmiques entre eux : disopyramide (Rythmodan®), quinidine, amiodarone (Cordarone®) et sotalol (Sotalex®) sont chacun allongeurs du QT. Leur association multiplie ce risque de façon non linéaire.
- Mizolastine (Mizollen®) + macrolides (érythromycine, clarithromycine, télithromycine) : double allongement du QT, association contre-indiquée. Les antihistaminiques H1 de deuxième génération non sédatifs comme la cétirizine ou la loratadine ne partagent pas ce risque.
Sulfamides hypoglycémiants et miconazole : une hypoglycémie sournoise
Le miconazole (Daktarin® gel buccal ou voie générale) est un antifongique azolé — inhibiteur puissant du CYP2C9, l’enzyme qui dégrade les sulfamides hypoglycémiants (glibenclamide Daonil®, gliclazide Diamicron®, glimépiride Amarel®, glipizide Minidiab®). La concentration du sulfamide monte en flèche, avec un risque de coma hypoglycémique. Cette interaction est d’autant plus piégieuse que le Daktarin® gel buccal est vendu sans ordonnance.
⚠️ Daktarin® gel buccal + diabétique sous sulfamides : refus systématique
Même en gel buccal, une fraction du miconazole est résorbée par la muqueuse orale. Chez un patient diabétique sous sulfamide, cette interaction est contre-indiquée. Orienter vers l’amphotéricine B (Fungizone® suspension buvable) ou la nystatine, antifongiques polyéniques non résorbés par le tube digestif, sans interaction métabolique.
Tramadol, ISRS et syndrome sérotoninergique : la triade à ne pas sous-estimer
Le syndrome sérotoninergique résulte d’un excès de sérotonine dans les synapses du système nerveux central. Il est souvent la conséquence d’une interaction médicamenteuse non reconnue. Une étude française portant sur 125 patients présentant ce syndrome a montré que dans 60 % des cas, il s’agissait d’une interaction médicamenteuse, principalement entre le tramadol et un inhibiteur sélectif de la recapture de la sérotonine (ISRS).
Une analyse d’études publiée en 2024 mentionne que l’incidence du syndrome sérotoninergique après usage de doses thérapeutiques varie de 0,006 % à 25 % selon les combinaisons, avec des taux particulièrement élevés en cas de surdosage : 15 % après surdosage d’ISRS ou d’IRSN, et jusqu’à 55 % après intoxication au moclobémide associé à d’autres agents sérotoninergiques.
Les combinaisons à risque comprennent : tramadol + ISRS (fluoxétine, paroxétine, sertraline, escitalopram), tramadol + IMAO (même sélectif de type A comme le moclobémide Moclamine®), tramadol + triptans, lithium + ISRS, et millepertuis + ISRS.
🔑 Reconnaître le syndrome sérotoninergique
La triade classique associe des signes neurovégétatifs (hyperthermie, sueurs, tachycardie, hypertension), neuromusculaires (tremblement, myoclonies, hyperréflexie, incoordination motrice) et neuropsychiques (agitation, confusion, troubles de conscience). Les symptômes débutent typiquement dans les 24 heures suivant la mise en place de la combinaison responsable. C’est une urgence médicale.
Sympathomimétiques et vasoconstriction additive
Les décongestionnants oraux — pseudoéphédrine (Rhinadvil®, Sudafed®) et phényléphrine (Hexarhume®) — associés aux vasoconstricteurs nasaux (naphazoline Derinox®, oxymétazoline Aturgyl®, tuaminoheptane Rhinofluimucil®) peuvent provoquer une sommation de leurs effets vasoconstricteurs artériels. La conséquence : hypertension paroxystique, dans des cas rares accidents vasculaires cérébraux, crises angineuses ou infarctus. Ces associations sont déconseillées, particulièrement chez les patients hypertendus ou cardiaques.
👨⚕️ Conseil au comptoir
En pratique, la co-dispensation d’un décongestionnant oral et d’un spray nasal vasoconstricteur lors d’un rhume est fréquente. Toujours vérifier si le patient prend déjà l’un des deux avant de proposer l’autre, et interroger systématiquement sur l’hypertension et les antécédents cardiovasculaires.
6. Anticoagulants oraux directs (AOD) : un profil d’interactions médicamenteuses à part
Les anticoagulants oraux directs (AOD) — dabigatran (Pradaxa®, inhibiteur direct de la thrombine), rivaroxaban (Xarelto®), apixaban (Eliquis®) et édoxaban (Lixiana®, inhibiteurs du facteur Xa) — ont largement remplacé les AVK dans de nombreuses indications depuis leur autorisation entre 2008 et 2015. Contrairement aux AVK, les AOD produisent un effet anticoagulant plus prévisible et moins variable, sans nécessiter de surveillance biologique régulière de l’INR.
Leur métabolisme fait surtout intervenir le CYP3A4 (pour le rivaroxaban et l’apixaban) et la P-glycoprotéine (pour les quatre molécules). Les données pharmacocinétiques suggèrent un risque d’interactions moindre avec l’apixaban comparativement aux autres AOD, ce qui est soutenu par des études de biodisponibilité et d’élimination rénale.
| Médicament associé | Mécanisme | Conséquence | Niveau ANSM | Preuve ℹ️ |
|---|---|---|---|---|
| Rifampicine | Inducteur puissant CYP3A4 + P-gp | Concentration AOD divisée par 2 à 4 → risque thrombotique | CI | ⭐⭐⭐⭐⭐ |
| Millepertuis | Inducteur CYP3A4 + P-gp | Diminution efficacité anticoagulante | CI | ⭐⭐⭐⭐ |
| Antifongiques azolés (fluconazole, kétoconazole) | Inhibiteur puissant CYP3A4 + P-gp | ↑ concentration AOD → risque hémorragique | AD à CI | ⭐⭐⭐⭐ |
| AINS / Aspirine ≥ 325 mg | Addition pharmacodynamique sur l’hémostase | Majoration du risque hémorragique digestif | AD | ⭐⭐⭐⭐⭐ |
| Ciclosporine | Inhibition forte P-gp | ↑ biodisponibilité AOD | CI (dabigatran) / PE (autres) | ⭐⭐⭐ |
⚠️ AOD et antitumoraux : une vigilance renforcée
Les prescriptions concomitantes d’AOD et de traitements anticancéreux sont de plus en plus fréquentes avec l’essor de la médecine ambulatoire en oncologie. Une analyse publiée dans Le Pharmacien Clinicien en 2024, portant sur 121 traitements antitumoraux, a mis en évidence des interactions potentiellement significatives avec les AOD pour une proportion importante de ces molécules. En oncologie, une évaluation pharmaceutique systématique avant toute introduction d’AOD est recommandée.
👨⚕️ Conseil au comptoir
Un patient sous AOD qui vous demande un antifongique vaginal (éconazole, clotrimazole) ou buccal local n’a aucun risque d’interaction : ces formes topiques ne sont pas résorbées systématiquement. En revanche, un traitement antifongique oral par fluconazole même en dose unique (Triflucan® 150 mg) est une association déconseillée avec les AOD. Toujours alerter le prescripteur.
7. Millepertuis : la plante aux cent interactions médicamenteuses
L’Hypericum perforatum (millepertuis) est la star incontestée des interactions plantes-médicaments. Utilisé pour ses propriétés antidépressives légères, il se comporte biochimiquement comme un véritable inducteur enzymatique polyvalent. Ses interactions pharmacocinétiques résultent d’une induction simultanée de plusieurs isoenzymes du cytochrome P450 — CYP3A4, CYP2C9, CYP2C19 — et de la P-glycoprotéine. L’induction enzymatique peut persister pendant une semaine après l’arrêt du millepertuis.
Le Thésaurus des interactions médicamenteuses de l’ANSM (édition septembre 2023) répertorie plusieurs contre-indications absolues avec le millepertuis. En cas d’association fortuite avec l’un de ces traitements, le millepertuis ne doit pas être arrêté de manière brutale : la suppression soudaine de l’effet inducteur expose le patient à un surdosage rapide des médicaments à marge thérapeutique étroite, notamment les anticoagulants de type AVK.
| Classe thérapeutique | Exemples de médicaments | Conséquence de l’interaction | Niveau | Preuve ℹ️ |
|---|---|---|---|---|
| Anticoagulants AVK | Warfarine, fluindione (Previscan®), acénocoumarol | Chute de l’INR → risque thrombotique (AVC, embolie) | CI | ⭐⭐⭐⭐⭐ |
| Anticoagulants oraux directs (AOD) | Rivaroxaban, apixaban, dabigatran | ↓ concentration plasmatique → perte d’efficacité anticoagulante | CI | ⭐⭐⭐⭐ |
| Immunosuppresseurs | Ciclosporine, tacrolimus (transplantés) | Concentration effondrée → rejet de greffe | CI | ⭐⭐⭐⭐⭐ |
| Contraceptifs hormonaux | Pilules œstroprogestatives, progestatives seules | Augmentation du métabolisme hépatique des estrogènes et des progestatifs → risque de grossesse non planifiée | CI | ⭐⭐⭐⭐ |
| Antiépileptiques | Carbamazépine, acide valproïque, lamotrigine… | ↓ concentrations plasmatiques → risque de crises | CI | ⭐⭐⭐⭐ |
| ISRS / Antidépresseurs | Fluoxétine, paroxétine, sertraline | Risque de syndrome sérotoninergique (action sérotoninergique additive) | CI | ⭐⭐⭐ |
| Antirétroviraux | Inhibiteurs de protéase, INNTI | Risque d’échec virologique → résistances VIH | CI | ⭐⭐⭐⭐ |
🚫 Millepertuis : une plante « naturelle » mais à risque élevé
« Naturel » ne signifie pas « sans danger ». Le millepertuis est l’une des plantes les plus richement documentées en termes d’interactions médicamenteuses graves. Si un patient vous présente un produit à base de millepertuis, la première question est : « Prenez-vous un traitement régulier ? » La réponse orientera presque toujours vers une contre-indication ou une très forte précaution d’emploi. Référer systématiquement au médecin prescripteur avant toute délivrance.
👨⚕️ Conseil au comptoir
N’arrêtez jamais le millepertuis brutalement chez un patient sous AVK : l’induction disparaissant progressivement, l’INR peut remonter dangereusement dans les jours qui suivent, avec un risque hémorragique. Le sevrage doit être progressif, sous contrôle de l’INR rapproché, en coordination avec le médecin traitant.
8. Prévenir les interactions médicamenteuses : les réflexes qui sauvent
La prévention des interactions médicamenteuses repose moins sur la mémorisation exhaustive des combinaisons à risque que sur quelques réflexes systématiques et des outils fiables. Voici ce qui fait vraiment la différence en pratique.
Les outils de référence
- Thésaurus ANSM des interactions médicamenteuses : la référence nationale française, mise à jour régulièrement (dernière révision majeure : septembre 2023). Gratuit, exhaustif, avec niveaux de contrainte explicites.
- Base de données publique des médicaments : les RCP (Résumés des Caractéristiques du Produit) de chaque médicament détaillent les interactions connues dans la rubrique « Interactions médicamenteuses ».
- Le Dossier Pharmaceutique (DP) : outil numérique officiel qui recense les dispensations des 21 derniers mois dans toutes les pharmacies françaises. Indispensable pour détecter les automédications non déclarées et les prescriptions parallèles. Si vous n’en avez pas encore un, demandez à votre pharmacien de l’ouvrir.
Les cinq questions qui changent tout
- « Prenez-vous d’autres médicaments en dehors de cette ordonnance ? » — y compris les médicaments sans ordonnance, les vitamines, les compléments alimentaires et les plantes médicinales.
- « Avez-vous consulté d’autres médecins récemment ? » — pour détecter les prescriptions parallèles non coordonnées.
- « Avez-vous changé votre alimentation ? » — le jus de pamplemousse (inhibiteur CYP3A4 intestinal), la vitamine K alimentaire (AVK), le soja (interactions thyroïdiennes) peuvent tous modifier l’efficacité d’un traitement.
- « Prenez-vous des plantes ou des produits naturels ? » — millepertuis, ginkgo biloba, ail concentré, échinacea… Les interactions phytothérapeutiques sont sous-estimées et sous-déclarées.
- « Avez-vous noté de nouveaux symptômes depuis le début du traitement ? » — certaines interactions se révèlent cliniquement avant d’être identifiées biologiquement.
ℹ️ Méthadone et interactions : alerte de l’ANSM (mai 2026)
En mai 2026, l’ANSM a rappelé le bon usage de la méthadone pour réduire le risque de surdose et d’interactions médicamenteuses, s’appuyant sur trois enquêtes nationales de vigilance menées entre 2022 et 2025 qui signalent une augmentation des cas. La méthadone est un substrat du CYP3A4 et du CYP2D6, avec une marge thérapeutique étroite : toute introduction d’un inhibiteur ou d’un inducteur enzymatique dans ce contexte doit faire l’objet d’une réévaluation médicale immédiate.
Tableau récapitulatif des principales interactions médicamenteuses à connaître
| Association à risque | Mécanisme | Risque clinique | Niveau | Preuve ℹ️ |
|---|---|---|---|---|
| Ergotés + macrolides | Inhibition CYP3A4 → accumulation ergotés | Ergotisme, ischémie | CI | ⭐⭐⭐⭐⭐ |
| Ergotés + triptans | Addition vasoconstrictrice | Ischémie cardiaque/cérébrale | CI | ⭐⭐⭐⭐⭐ |
| Antiparkinsoniens + métoclopramide | Antagonisme récepteurs D2 | Aggravation syndrome parkinsonien | CI | ⭐⭐⭐⭐⭐ |
| Millepertuis + AVK / AOD | Induction CYP3A4/2C9/P-gp | Perte d’efficacité anticoagulante | CI | ⭐⭐⭐⭐⭐ |
| Miconazole oral + sulfamides | Inhibition CYP2C9 | Hypoglycémie sévère, coma | CI | ⭐⭐⭐⭐⭐ |
| Tramadol + ISRS | Addition sérotoninergique | Syndrome sérotoninergique | AD | ⭐⭐⭐⭐ |
| Statines + fibrates | Toxicité musculaire additive | Rhabdomyolyse, IRA | AD | ⭐⭐⭐⭐⭐ |
| AOD + antifongiques azolés oraux | Inhibition CYP3A4 + P-gp | Surdosage anticoagulant, hémorragie | AD à CI | ⭐⭐⭐⭐ |
| AVK + AINS / Aspirine | Addition pharmacodynamique hémostatique | Hémorragie digestive, intracrânienne | AD | ⭐⭐⭐⭐⭐ |
| Pseudoéphédrine + vasoconstricteurs nasaux | Addition vasoconstrictrice | HTA, AVC, IDM (rares) | AD | ⭐⭐⭐ |
CI = Contre-indication ; AD = Association Déconseillée ; PE = Précaution d’Emploi. IRA = insuffisance rénale aiguë. Source : ANSM Thésaurus des interactions médicamenteuses, mise à jour juin 2024.
🔑 En résumé : interactions médicamenteuses, ce qu’il faut retenir
Les interactions médicamenteuses se produisent à deux niveaux : les interactions pharmacodynamiques (effets qui s’additionnent ou s’antagonisent sur le même récepteur ou système) et les interactions pharmacocinétiques (perturbation de l’absorption, du métabolisme — notamment par les cytochromes P450 — ou de l’élimination).
Les combinaisons les plus dangereuses à retenir en pratique officinale : ergotés + macrolides (ergotisme), antiparkinsoniens + métoclopramide (aggravation Parkinson), millepertuis + anticoagulants (perte d’efficacité), miconazole + sulfamides (hypoglycémie), tramadol + ISRS (syndrome sérotoninergique).
L’outil central du pharmacien est le Thésaurus ANSM (mis à jour en juin 2024) et le Dossier Pharmaceutique. Les cinq questions systématiques (autres médicaments, autres médecins, alimentation, plantes, nouveaux symptômes) permettent de détecter la majorité des situations à risque avant qu’elles ne deviennent cliniquement significatives.
🔗 Articles connexes sur Astuces Pharma
Avertissement médical : Cet article a une vocation informative et pédagogique. Il ne se substitue pas à l’avis d’un professionnel de santé. En cas de doute sur une interaction médicamenteuse, consultez systématiquement votre pharmacien ou votre médecin avant toute modification de traitement. Ne jamais arrêter ni modifier un traitement sans avis médical.
Sources principales : ANSM, Thésaurus des interactions médicamenteuses, mise à jour juin 2024 (ansm.sante.fr) · ANSM, Rapport d’activité 2024 · Deodhar M. et al., Pharmaceutics, 2020, DOI:10.3390/pharmaceutics12090846 · Centre Antipoisons de Belgique, Syndrome sérotoninergique, 2025 · CBIP Folia, Interactions contraceptifs hormonaux, 2024 · Le Pharmacien Clinicien, AOD et antitumoraux, Volume 59, 2024 · Revue Pharma, Millepertuis et interactions ANSM 2023, novembre 2024.
Article rédigé par Anne-Sophie DELEPOULLE (Dr en Pharmacie) — Dernière mise à jour : mai 2026